Publications
Selected Work by Topic
Physics-Informed Computational Design of Photoenzymes
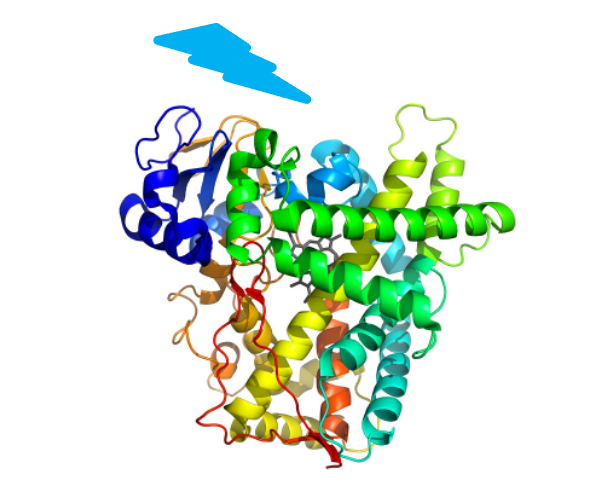
Certain natural enzymes can be repurposed by light and carry out non-natural reactions. These enzymes can be tuned through protein engineering to carry out reactions that are challenging for other types of catalysts. We develop design strategies and predictive tools based on first-principles simulations to advance the emerging field of photoenzymatic catalysis.
Bhumika Jayee, Sunny Lee, Sijia S. Dong*. Sequence-Transferrable Machine Learning Prediction of Flavin-Dependent Photoenzyme Spectral Properties. ChemRxiv. 2026. DOI: 10.26434/chemrxiv.15002532/v2
Gustavo Mondragón-Solórzano, Sijia S. Dong*. Can the photoinduced electron transfer in GluER photoenzymes occur through a triplet state? ChemRxiv. 2026. DOI: 10.26434/chemrxiv.15004891/v1
Felipe Curtolo, Sijia S. Dong*. Molecular Origins of Simultaneous Chemo-, Enantio-, and Substrate Selectivity in Non-Natural Photoenzymatic Radical Reactions. J. Am. Chem. Soc., 2025, 147, 45, 41639–41649.
Jose M. Carceller^, Bhumika Jayee^, Claire G. Page, Daniel G. Oblinsky, Gustavo Mondragón-Solórzano, Nithin Chintala, Jingzhe Cao, Zayed Alassad, Zheyu Zhang, Nathaniel White, Danny J. Diaz, Andrew D. Ellington, Gregory D. Scholes*, Sijia S. Dong*, Todd K. Hyster*. Engineering a Photoenzyme to Use Red Light. Chem. 2025, 11, 102318. | Highlighted in Preview: Spectral Control of Photoenzyme Through Allosteric Chromophore Tuning.
Computational Design of Polymer-Based Enzyme Mimics
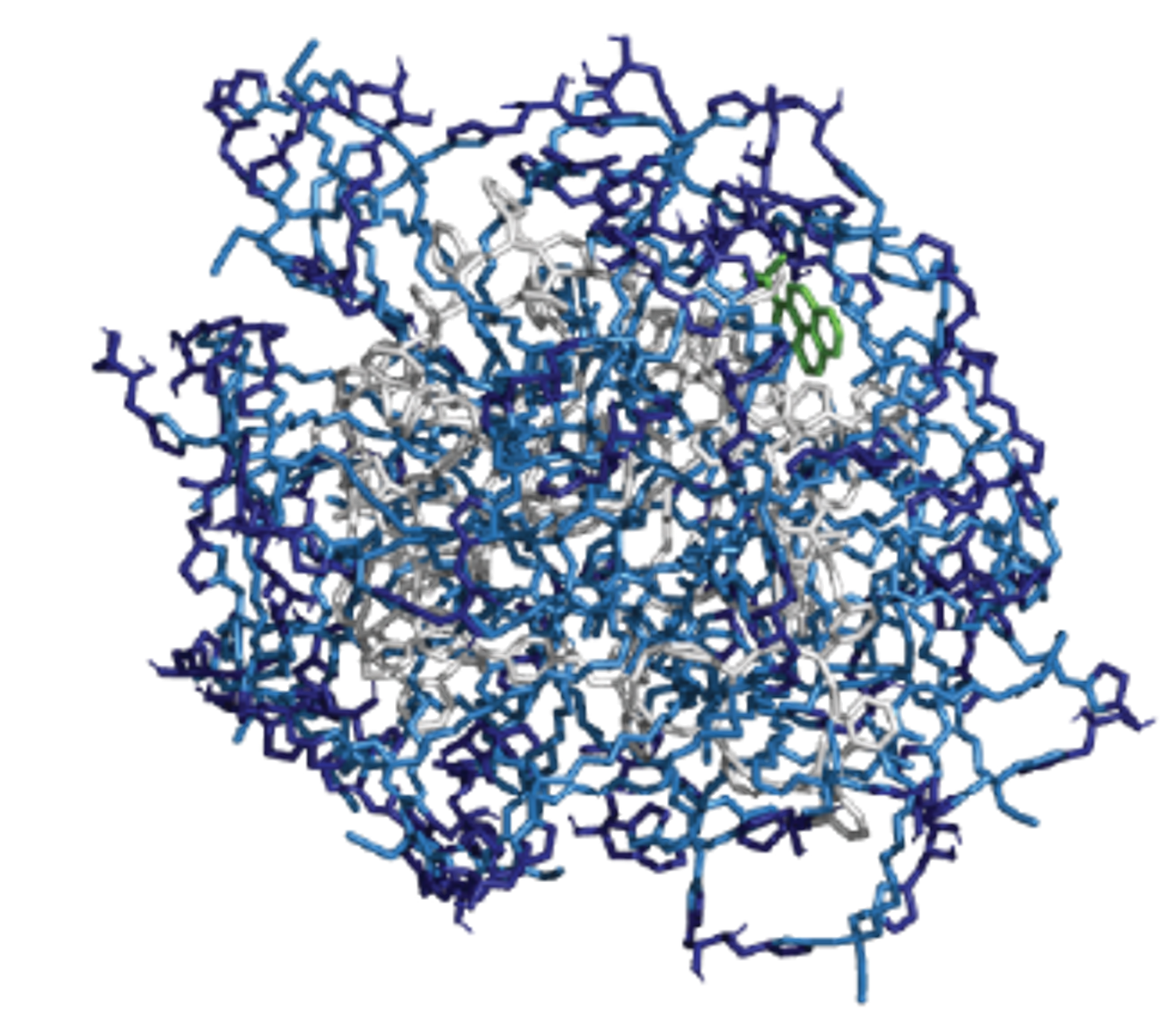
Synthetic polymers and molecular imprinting techniques can provide the scaffold of macromolecules that have protein-mimicking functionality such as catalysis and sensing but can endure a wider range of reaction conditions than proteins. We develop computational methods to model and design polymer-based enzyme mimics.
Emma Stevens, Felipe Curtolo, Bradford Derby, Dashaunette Wallace, Yan Zhao, Sijia S. Dong*. Modeling Molecularly Imprinted Nanoparticles with LNKD: A Resource Efficient Algorithm for Polymer Crosslinking. J. Chem. Inf. Model., 2025, 65, 19, 10408–10417.
Computational Chemistry on Quantum Computers
Quantum computing has the potential to revolutionize computational science. We develop algorithms towards large-scale quantum chemistry simulations on near-term quantum computers.
Bosonic Quantum Computing
We explore bosonic quantum computing for quantum chemistry applications. We introduce the qumode-based variational quantum deflation framework (QumVQD) for calculating both the electronic and vibrational excited states of molecules on qumode-based devices. We demonstrated bosonic quantum devices’ potential advantages over qubit-based devices regarding noise resilience and gate counts for computing vibrational states.
Marlon F. Jost, Sijia S. Dong*. Excited-State Quantum Chemistry on Qumode-Based Processors via Variational Quantum Deflation. arXiv. 2026. arXiv:2604.13457
Quantum Annealing
We introduce a variational quantum annealing (VarQA) algorithm for electronic structure theory, in which we use the quantum annealer as a sampler and prepare an ansatz state through its statistics. This method improves the scaling of the number of logical qubits from exponential to linear for electronic structure theory on quantum annealers. We demonstrated the effectiveness of the method on D-Wave's quantum hardware.
Ka-Wa Yip, Kubra Yeter-Aydeniz, Sijia S. Dong*. Variational Quantum Annealing for Quantum Chemistry. arXiv. 2025. arXiv:2503.15473
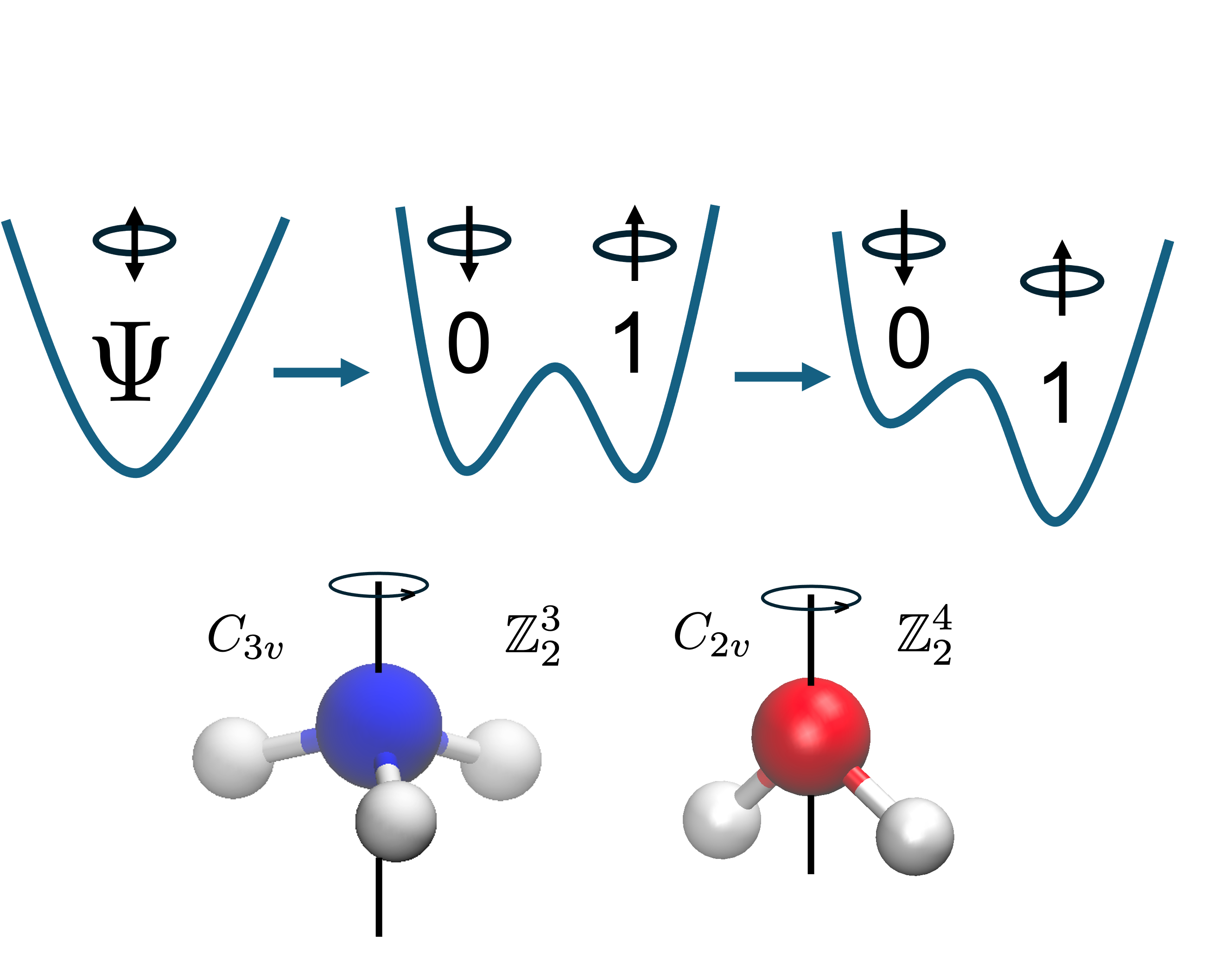
We reduced the qubit requirement for electronic structure theory on quantum annealers by using symmetry-adapted encodings. This provides an exponential reduction in the size of the Hilbert space. We are able to simulate molecules larger than those previously reported. We also provide an explanation for the errors of the method as related to the multireference character of the system, and briefly discuss the potential of extracting electronic excited states from our method.
Joseph W. Desroches, Sijia S. Dong*. Electronic Structure Theory with Molecular Point Group Symmetries on Quantum Annealers. J. Chem. Phys., 2025, 165, 244104. | 2024 JCP Emerging Investigators Special Collection
Accelerating Quantum Chemistry Simulations of Molecules and Condensed Systems: Machine Learning, Automation, and Other Computational Tools
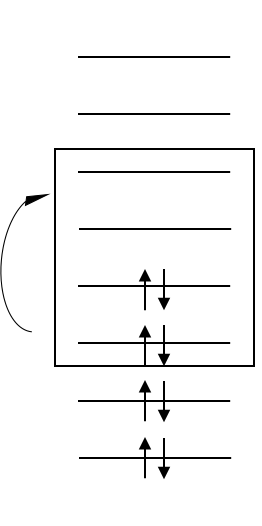
Multireference (Strongly correlated) systems are ubiquitous in nature and in new generations of materials. For example, accurate description of photochemistry and transition metal spin states often requires the proper treatment of strong correlation. To study such systems, it is common and often necessary to use multireference methods. However, multireference methods face computational challenges.
The procedures for choosing a starting point to optimize multiconfigurational wave functions is often nonsystematic or not optimal in their efficiency. We have developed several approaches to address the issue of generating reasonable reference wave functions for complete active space 2nd-order perturbation theory and multiconfiguration pair-density functional theory to accurately predict the excited states of various molecules. We also use machine learning to replace expensive multireference calculations.
Multireference calculations are computationally demanding and can be intractble. We have developed machine learning surrogate models that reduce the number of multireference calculations needed to predict the spectral properties of flavin-dependent photoenzymes.
Bhumika Jayee, Sunny Lee, Sijia S. Dong*. Sequence-Transferrable Machine Learning Prediction of Flavin-Dependent Photoenzyme Spectral Properties. ChemRxiv. 2026. DOI: 10.26434/chemrxiv.15002532/v2
We developed a set of procedures to construct potential energy surfaces using multireference methods in a parallel fashion. We demonstrated an order of magnitude saving in computing time for constructing the potential energy curves of diatomic molecules such as dichromium.
Benjamin W. Kaufold, Adithya Palle, Sijia S. Dong*. Parallel Procedures for Efficient Construction of Potential Energy Surfaces Based on Multireference Methods. ChemRxiv. 2026. DOI: 10.26434/chemrxiv-2025-j0vns/v2

We developed a scheme to use a physical observable, the dipole moment, to select the active space automatically. Our scheme can potentially accelerate the time-to-solution by ten times. We demonstrated the effectiveness of the method for calculating the vertical excitation energies of a collection of organic molecules, spin states of transition metal oxides, and bond dissociation curves of diatomic molecules.
Benjamin W. Kaufold, Sijia S. Dong*. Automated Active Space Selection with CASCI Dipole Moments. ChemRxiv. 2026. DOI: 10.26434/chemrxiv.15000921/v2
Benjamin W. Kaufold, Nithin Chintala, Pratima Pandeya, Sijia S. Dong*. Automated Active Space Selection with Dipole Moments. J. Chem. Theory Comput., 2023, 19, 9, 2469–2483.
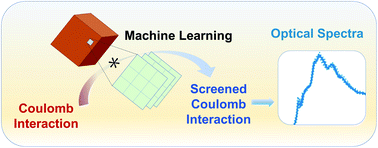
Accurate and efficient predictions of absorption spectra of molecules and solids are essential for the understanding and rational design of broad classes of materials, including photo-absorbers in solar and photo-electrochemical cells and defective insulators and semiconductors hosting optically addressable spin-defects. The prediction of absorption spectra at finite temperature is needed to directly compare with experimental observables. We have developed an approach to improve the efficiency of first principles calculations of absorption spectra of complex materials at finite temperature, based on the solution of the Bethe-Salpeter equation (BSE) and machine-learning techniques.
We demonstrate that methods based on convolution can be efficiently used to compute the screened Coulomb interaction, a key component of BSE calculations, with computational gains of one to two orders of magnitude for systems with 50 to 500 atoms, including liquids, solids, nanostructures, and solid/liquid interfaces. Importantly, our approach yields interpretable machine learning results, from which model dielectric functions may be derived. These model functions may be used not only in the BSE but also in developing functionals for time-dependent density functional theory (TDDFT) calculations, for both homogeneous and heterogeneous systems. Our work provides a strategy to combine machine learning with electronic structure calculations to accelerate the first principles simulations of excited-state properties.
Sijia S. Dong, Marco Govoni, Giulia Galli. Machine Learning Dielectric Screening for the Simulation of Excited State Properties of Molecules and Materials. Chem. Sci., 2021, 12, 4970-4980. | Editor’s Choice – Graeme Day | Materials & Energy in Chemical Science - most popular articles 2021
Click Here for More Selected Work by Topic
Publication List
(Preprint) Gustavo Mondragón-Solórzano, Sijia S. Dong*. Can the photoinduced electron transfer in GluER photoenzymes occur through a triplet state? ChemRxiv. 2026. DOI: 10.26434/chemrxiv.15004891/v1
(Preprint) Bhumika Jayee, Sunny Lee, Sijia S. Dong*. Sequence-Transferrable Machine Learning Prediction of Flavin-Dependent Photoenzyme Spectral Properties. ChemRxiv. 2026. DOI: 10.26434/chemrxiv.15002532/v2
(Preprint) Marlon F. Jost, Sijia S. Dong*. Excited-State Quantum Chemistry on Qumode-Based Processors via Variational Quantum Deflation. arXiv. 2026. arXiv:2604.13457
(Preprint) Benjamin W. Kaufold, Sijia S. Dong*. Automated Active Space Selection with CASCI Dipole Moments. ChemRxiv. 2026. DOI: 10.26434/chemrxiv.15000921/v2
(Preprint) Benjamin W. Kaufold, Adithya Palle, Sijia S. Dong*. Parallel Procedures for Efficient Construction of Potential Energy Surfaces Based on Multireference Methods. ChemRxiv. 2026. DOI: 10.26434/chemrxiv-2025-j0vns/v2
Sumana L. Raj, Felipe Curtolo, Kacie Nelson, David A. Cagan, Reagan X. Hooper, Daniel Bím, Alec H. Follmer, Ryan D. Ribson, Nathanael P. Kazmierczak, Brendon J. McNicholas, Natalia Powers-Riggs, Michael Sachs, Elisa Biasin, Dimosthenis Sokaras, Jinkyu Lim, Leland B. Gee, Matthieu Chollet, Patrick Kramer, Roberto Alonso Mori, Tim B. van Driel, Kelly Gaffney, Sijia S. Dong*, Ryan G. Hadt, and Amy A. Cordones*. Ultrafast Population and Structural Dynamics of a Ni-Bipyridine Photoredox Catalyst Reveal a Significant Deactivation Pathway. J. Phys. Chem. Lett. 2026. DOI: 10.1021/acs.jpclett.5c03264
(Editorial) Zhongyue John Yang*, Sijia S. Dong*. Modeling the Impact of Protein Mutations on Chemical Reactions for Biochemistry, Protein Engineering, and Drug Discovery. J. Chem. Theory Comput. 2026. DOI: 10.1021/acs.jctc.5c01912
(Perspective) Ferguson, A.; LaFleur, M.; Ruthotto, L.; Thaler, J.; Ting, Y.-S.; Tiwary, P.; Villar, S.; Alves, E. P.; Avigad, J.; Billinge, S.; Bilodeau, C.; Brown, K.; Candes, E.; Chattopadhyay, A.; Cheng, B.; Clausen, J.; Coley, C.; Connolly, A.; Daum, F.; Dong, S.; Du, C. X.; Dvorkin, C.; Fanelli, C.; Ford, E. B.; Frutos, L. M.; Trillos, N. G.; Garraffo, C.; Ghrist, R.; Gomez-Bombarelli, R.; Guadagni, G.; Guggilam, S.; Gukov, S.; Gutiérrez, J. B.; Habib, S.; Hachmann, J.; Hanin, B.; Harris, P.; Holland, M.; Holm, E.; Huang, H.-Y.; Hsu, S.-C.; Jackson, N.; Isayev, O.; Ji, H.; Katsaggelos, A.; Kepner, J.; Kevrekidis, Y.; Kuchera, M.; Kutz, J. N.; Lalic, B.; Lee, A.; LeBlanc, M.; Lim, J.; Lindsey, R.; Liu, Y.; Lu, P. Y.; Malik, S.; Mandic, V.; Manian, V.; Mazi, E. P.; Mehta, P.; Melchior, P.; Ménard, B.; Ngadiuba, J.; Offner, S.; Olivetti, E.; Ong, S. P.; Rackauckas, C.; Rigollet, P.; Risko, C.; Romero, P.; Rotskoff, G.; Savoie, B.; Seljak, U.; Shih, D.; Shiu, G.; Shlyakhtenko, D.; Silverstein, E.; Sparks, T.; Strohmer, T.; Stubbs, C.; Thomas, S.; Vaikuntanathan, S.; Vidal, R.; Villaescusa-Navarro, F.; Voth, G.; Wandelt, B.; Ward, R.; Weber, M.; Wechsler, R.; Whitelam, S.; Wiest, O.; Williams, M.; Yang, Z.; Yingling, Y. G.; Yu, B.; Yue, S.; Zabludoff, A.; Zhao, H.; Zhang, T. The Future of Artificial Intelligence and the Mathematical and Physical Sciences (AI+MPS). arXiv. 2025. arXiv.2509.02661
Felipe Curtolo, Sijia S. Dong*. Molecular Origins of Simultaneous Chemo-, Enantio-, and Substrate Selectivity in Non-Natural Photoenzymatic Radical Reactions. J. Am. Chem. Soc., 2025, 147, 45, 41639–41649.
Emma Stevens, Felipe Curtolo, Bradford Derby, Dashaunette Wallace, Yan Zhao, Sijia S. Dong*. Modeling Molecularly Imprinted Nanoparticles with LNKD: A Resource Efficient Algorithm for Polymer Crosslinking. J. Chem. Inf. Model., 2025, 65, 19, 10408–10417.
Benjamin W. Kaufold, Parisa Nematollahi*, Bernardo Barbiellini, Dirk Lamoen, Arun Bansil, Hana Cheng, Sijia S. Dong*, Sanjeev Mukerjee*. Plasmon-Induced Resonant Energy Transfer and Flat Band Formation in Fe and Co Doped Ni(II) Hydroxide for Efficient Photocatalytic Oxygen Evolution. Phys. Chem. Chem. Phys., 2025, 27, 18015-18026.
(Preprint) Ka-Wa Yip, Kubra Yeter-Aydeniz, Sijia S. Dong*. Variational Quantum Annealing for Quantum Chemistry. arXiv. 2025. arXiv:2503.15473
Joseph W. Desroches, Sijia S. Dong*. Electronic Structure Theory with Molecular Point Group Symmetries on Quantum Annealers. J. Chem. Phys., 2025, 165, 244104. | 2024 JCP Emerging Investigators Special Collection
Jose M. Carceller^, Bhumika Jayee^, Claire G. Page, Daniel G. Oblinsky, Gustavo Mondragón-Solórzano, Nithin Chintala, Jingzhe Cao, Zayed Alassad, Zheyu Zhang, Nathaniel White, Danny J. Diaz, Andrew D. Ellington, Gregory D. Scholes*, Sijia S. Dong*, Todd K. Hyster*. Engineering a Photoenzyme to Use Red Light. Chem. 2025, 11, 102318. | Highlighted in Preview: Spectral Control of Photoenzyme Through Allosteric Chromophore Tuning.
(Editorial) Burton, H. G. A.; Dong, S. S.*; Ghosh, S.; Gu, B.; Jackson, N. E.; Keefer, D.; Lu, Y.; Monroe, J. I.; Peng, B.; Pieri, E.; Spackman, P. R.; Vacher, M.; Vuckovic, S.; Williams-Young, D.; Yang, Z. J.; Yue, S.; Zerze, G. H.; Zhu, T. JCTC Early Career Board Selects. J. Chem. Theory Comput. 2024, 20 (14), 5785–5787.
Katherine Carney, Lauren Polito, Kamilya Reid, Surbhi Srinivas, Gabrielle Blake, Nithin Chintala, Sijia S. Dong* and Rein Kirss*. Kinetics and Mechanism of Halide Exchange in Reactions of CpRu(PPh3)2Cl with Alkyl Halides: Evidence for Radical Pairs. New J. Chem., 2023,47, 21404-21410.
Benjamin W. Kaufold, Nithin Chintala, Pratima Pandeya, Sijia S. Dong*. Automated Active Space Selection with Dipole Moments. J. Chem. Theory Comput., 2023, 19, 9, 2469–2483.
Li Manni, G.; Fdez. Galván, I.; Alavi, A.; Aleotti, F.; Aquilante, F.; Autschbach, J.; Avagliano, D.; Baiardi, A.; Bao, J. J.; Battaglia, S.; Birnoschi, L.; Blanco-González, A.; Bokarev, S. I.; Broer, R.; Cacciari, R.; Calio, P. B.; Carlson, R. K.; Carvalho Couto, R.; Cerdán, L.; Chibotaru, L. F.; Chilton, N. F.; Church, J. R.; Conti, I.; Coriani, S.; Cuéllar-Zuquin, J.; Daoud, R. E.; Dattani, N.; Decleva, P.; de Graaf, C.; Delcey, M. G.; De Vico, L.; Dobrautz, W.; Dong, S. S.; Feng, R.; Ferré, N.; Filatov(Gulak), M.; Gagliardi, L.; Garavelli, M.; González, L.; Guan, Y.; Guo, M.; Hennefarth, M. R.; Hermes, M. R.; Hoyer, C. E.; Huix-Rotllant, M.; Jaiswal, V. K.; Kaiser, A.; Kaliakin, D. S.; Khamesian, M.; King, D. S.; Kochetov, V.; Krośnicki, M.; Kumaar, A. A.; Larsson, E. D.; Lehtola, S.; Lepetit, M.-B.; Lischka, H.; López Ríos, P.; Lundberg, M.; Ma, D.; Mai, S.; Marquetand, P.; Merritt, I. C. D.; Montorsi, F.; Mörchen, M.; Nenov, A.; Nguyen, V. H. A.; Nishimoto, Y.; Oakley, M. S.; Olivucci, M.; Oppel, M.; Padula, D.; Pandharkar, R.; Phung, Q. M.; Plasser, F.; Raggi, G.; Rebolini, E.; Reiher, M.; Rivalta, I.; Roca-Sanjuán, D.; Romig, T.; Safari, A. A.; Sánchez-Mansilla, A.; Sand, A. M.; Schapiro, I.; Scott, T. R.; Segarra-Martí, J.; Segatta, F.; Sergentu, D.-C.; Sharma, P.; Shepard, R.; Shu, Y.; Staab, J. K.; Straatsma, T. P.; Sørensen, L. K.; Tenorio, B. N. C.; Truhlar, D. G.; Ungur, L.; Vacher, M.; Veryazov, V.; Voß, T. A.; Weser, O.; Wu, D.; Yang, X.; Yarkony, D.; Zhou, C.; Zobel, J. P.; Lindh, R. The OpenMolcas Web: A Community-Driven Approach to Advancing Computational Chemistry. J. Chem. Theory Comput., 2023, 19, 20, 6933–6991.
Lei Zhang^, Yuyan Wang^, Peiru Chen, Dali Wang, Tingyu Sun, Zheyu Zhang, Ruimeng Wang, Xi Kang, Yang Fang, Hao Lu, Jiansong Cai, Mengqi Ren, Sijia S. Dong*, and Ke Zhang*. A mechanistic study on the cellular uptake, intracellular trafficking, and antisense gene regulation of bottlebrush polymer-conjugated oligonucleotides. RSC Chem. Biol., 2023, 4, 138-145.
Yuyan Wang, Dali Wang, Jiachen Lin, Zidi Lyu, Peiru Chen, Tingyu Sun, Mehrnaz Mojtabavi, Armin Vedadghavami, Zheyu Zhang, Ruimeng Wang, Lei Zhang, Christopher Park, Sijia S. Dong*, and Ke Zhang*. A Long-Circulating Vector for Aptamers Based upon Polyphosphodiester-Backboned Molecular Brushes. Angew. Chem. Int. Ed., 2022, 61, e202204576.
Dali Wang, Qiwei Wang, Yuyan Wang, Peiru Chen, Xueguang Lu, Fei Jia, Yehui Sun, Tingyu Sun, Lei Zhang, Fangyuan Che, Jialu He, Liming Lian, Gemma Morano, Michael Shen, Mengqi Ren, Sijia S. Dong, Jean Zhao, and Ke Zhang. Targeting oncogenic KRAS with molecular brush-conjugated antisense oligonucleotides. PNAS, 2022, 119 (29), e2113180119.
Benjamin S. Rich, Noah B. Bissonnette, Alejandra Duran Balsa, Mulan Yang, Hope Meikle, Nithin Chintala, Sijia S. Dong* and Rein U. Kirss*. Mechanism of Halide Exchange in Reactions of CpRu(PPh3)2Cl with Haloalkanes. New J. Chem., 2022, 46, 6603-6608.
Sijia S. Dong, Marco Govoni, Giulia Galli. Machine Learning Dielectric Screening for the Simulation of Excited State Properties of Molecules and Materials. Chem. Sci., 2021, 12, 4970-4980. | Editor’s Choice – Graeme Day | Materials & Energy in Chemical Science - most popular articles 2021
Before 2021
Ignacio F. Galván, Morgane Vacher, Ali Alavi, Celestino Angeli, Jochen Autschbach, Jie J. Bao, Sergey I. Bokarev, Nikolay A. Bogdanov, Rebecca K. Carlson, Liviu F. Chibotaru, Joel Creutzberg, Nike Dattani, Mickaël G. Delcey, Sijia S. Dong, Andreas Dreuw, Leon Freitag, Luis Manuel Frutos, Laura Gagliardi, Frédéric Gendron, Angelo Giussani, Leticia Gonzalez, Gilbert Grell, Meiyuan Guo, Chad E. Hoyer, Marcus Johansson, Sebastian Keller, Stefan Knecht, Goran Kovačević, Erik Källman, Giovanni Li Manni, Marcus Lundberg, Yingjin Ma, Sebastian Mai, João Pedro Malhado, Per Åke Malmqvist, Philipp Marquetand, Stefanie A. Mewes, Jesper Norell, Massimo Olivucci, Markus Oppel, Quan Manh Phung, Kristine Pierloot, Felix Plasser, Markus Reiher, Andrew M. Sand, Igor Schapiro, Prachi Sharma, Christopher J. Stein, Lasse Kragh Sørensen, Donald G. Truhlar, Mihkel Ugandi, Liviu Ungur, Alessio Valentini, Steven Vancoillie, Valera Veryazov, Oskar Weser, Per-Olof Widmark, Sebastian Wouters, J. Patrick Zobel, Roland Lindh. OpenMolcas: From Source Code to Insight. J. Chem. Theory Comput., 2019, 15, 11, 5925-5964. (Link)
Sijia S. Dong, Laura Gagliardi, Donald G. Truhlar. Nature of the \(1^1B_{u}\) and \(2^1A_{g}\) Excited States of Butadiene and the Goldilocks Principle of Basis Set Diffuseness. J. Chem. Theory Comput., 2019, 15, 8, 4591-4601. (Link)
Sijia S. Dong^, Benchen Huang^, Laura Gagliardi, Donald G. Truhlar. State-Interaction Pair-Density Functional Theory Can Accurately Describe a Spiro Mixed-Valence Compound. J. Phys. Chem. A, 2019, 123, 10, 2100-2106. (Published as part of The Journal of Physical Chemistry virtual special issue “Leo Radom Festschrift”.) (Link)
María del Carmen Marín, Luca De Vico, Sijia S. Dong, Laura Gagliardi, Donald G. Truhlar, Massimo Olivucci. Assessment of MC-PDFT Excitation Energies for a Set of QM/MM Models of Rhodopsins. J. Chem. Theory Comput., 2019, 15, 3, 1915-1923. (Link)
Yinan Shu, Sijia S. Dong, Kelsey A. Parker, Junwei L. Bao, Linyao Zhang, Donald G. Truhlar. Extended Hamiltonian Molecular Dynamics: Semiclassical Trajectories with Improved Maintenance of Zero Point Energy. Phys. Chem. Chem. Phys., 2018, 20, 30209-30218. (Link) | Highlighted as a 2018 PCCP HOT Article
Jie J. Bao, Sijia S. Dong, Laura Gagliardi, and Donald G. Truhlar. Automatic Selection of an Active Space for Calculating Electronic Excitation Spectra by MS-CASPT2 or MC-PDFT. J. Chem. Theory Comput., 2018, 14 (4), 2017–2025. (Link)
Sijia S. Dong, Laura Gagliardi, Donald G. Truhlar. Excitation Spectra of Retinal by Multiconfiguration Pair-Density Functional Theory. Phys. Chem. Chem. Phys., 2018, 20, 7265-7276. (Link)
(Book Chapter) Sijia S. Dong, William A. Goddard III, Ravinder Abrol. Identifying Multiple Active Conformations in the G Protein-Coupled Receptor Activation Landscape Using Computational Methods. In Methods in Cell Biology. 2017; vol. 142, pp. 173-186.(Link)
Sijia S. Dong, William A. Goddard III, Ravinder Abrol. Conformational and Thermodynamic Landscape of GPCR Activation from Theory and Computation. Biophys. J., 2016, 110 (12), 2618-2629. (Link)
Ruijie D. Teo^, Sijia S. Dong^, Zeev Gross, Harry B. Gray, William A. Goddard III. Computational Predictions of Corroles as a Class of Hsp90 Inhibitors. Molecular BioSystems, 2015, 11, 2907-2914. (Link)
Sijia S. Dong, Ravinder Abrol, William A. Goddard III. The Predicted Ensemble of Low Energy Conformations of Human Somatostatin Receptor Subtype 5 and the Binding of Antagonists. ChemMedChem, 2015, 10 (4), 650–661. (Link)
Xin-Yuan Liu^, Zhen Guo^, Sijia S. Dong^, Xiao-Hua Li and Chi-Ming Che. Highly Efficient and Diastereoselective Gold (I)-Catalyzed Synthesis of Tertiary Amines from Secondary Amines and Alkynes: Substrate Scope and Mechanistic Insights. Chemistry-A European Journal, 2011, 17 (46), 12932–12945. (Link)
Sijia S. Dong, Robert J. Nielsen, Joshua H. Palmer, Harry B. Gray, Zeev Gross, Siddharth Dasgupta, William A. Goddard III. Electronic Structures of Group 9 Metallocorroles with Axial Ammines. Inorganic Chemistry, 2011, 50 (3), 764–770. (Link)
^ denotes equal contribution